Go

When I presented a poster about SciCommander at the Swedish bioinformatics workshop last year, I got a lot of awesome feedback from some great people including Fredrik Boulund, Johannes Alneberg and others, of which I unfortunately lost the names (please shout out if you read this!).
(For those new to SciCommander, it …

As we are - according to some expert opinions - living in the Century of Biology, I found it interesting to reflect on Go’s usage within the field.
Go has some great features that make it really well suited for biology, such as:
A relatively simple language that can be learned in a short time even for people …

I have been playing around a lot with concurrency in Go over the years, resulting in libraries such as SciPipe , FlowBase and rdf2smw . My main motivation for looking into Go has been the possibility to use it as a more performant, scaleable and type-safe alternative to Python for data heavy scripting tasks in …
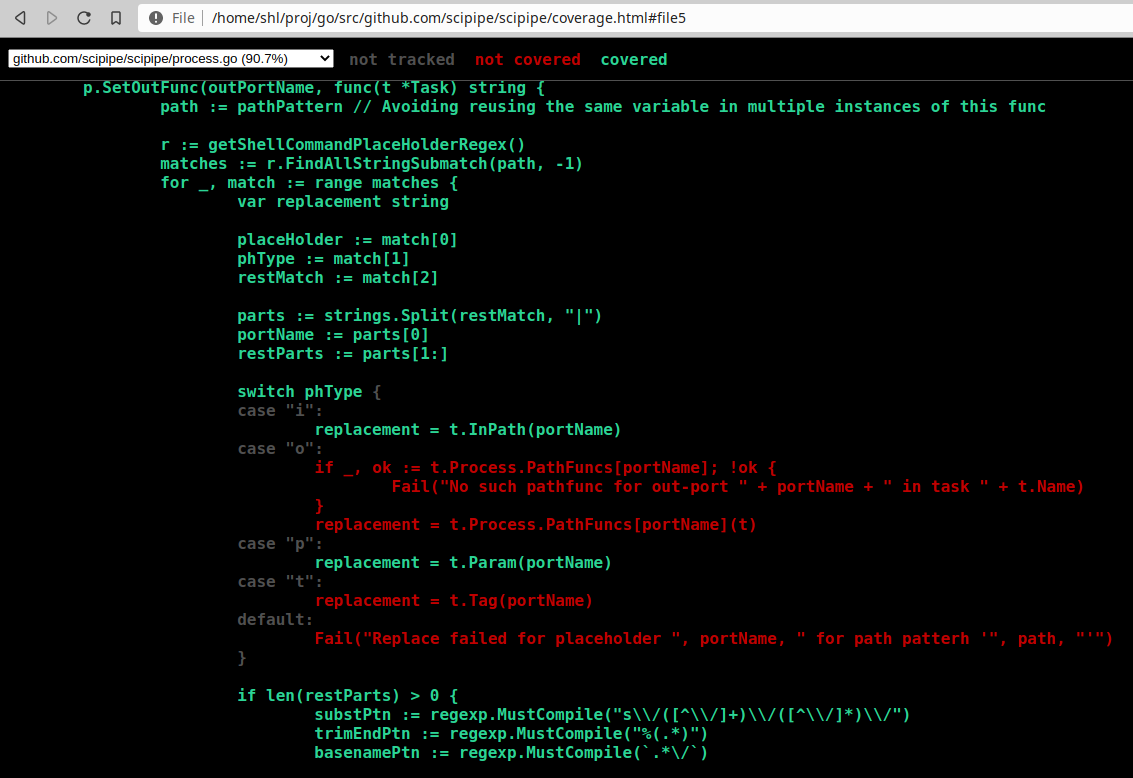
Go has some really nice tools for running tests and analyzing code. One of these functionalities is that you can generate coverage information when running tests, that can later be viewed in a browser using the go tool cover command. It turns out though, since doing it requires executing multiple commands after each …

I was so happy the other day to find someone else who found the great benefits of a little pattern for how to structure pipeline-heavy programs in Go, which I described in a few posts before. I have been surprised to not find more people using this kind of pattern, which has been so extremely helpful to us, so I …
I had a problem in which I thought I needed to parse the full DrugBank dataset, which comes as a (670MB) XML file (For open access papers describing DrugBank, see: [1], [2], [3] and [4]). It turned out what I needed was available as CSV files under “Structure External Links ”. There is probably still some …
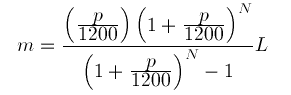
Mathematical notation and dataflow programming Even though computations done on computers are very often based on some type of math, it is striking that the notation used in math to express equations and relations is not always very readily converted into programming code. Outside of purely symbolic programming …

TL;DR: We wrote a post on gopherdata.io, about the growing ecosystem of Go-based workflow tools in bioinformatics. Go read it here It is interesting to note how Google’s Go programming language seems to increase in popularity in bioinformatics.
Just to give a sample of some of the Go based bioinformatics tools …
Thus, optimally, one would want to use Go’s handy range keyword for looping over multiple channels, since range takes care of closing the for-loop at the right time (when the inbound channel is closed). So something like this (N.B: non-working code!):
for a, b, c := range chA, chB, chC { doSomething(a, b, c) } …
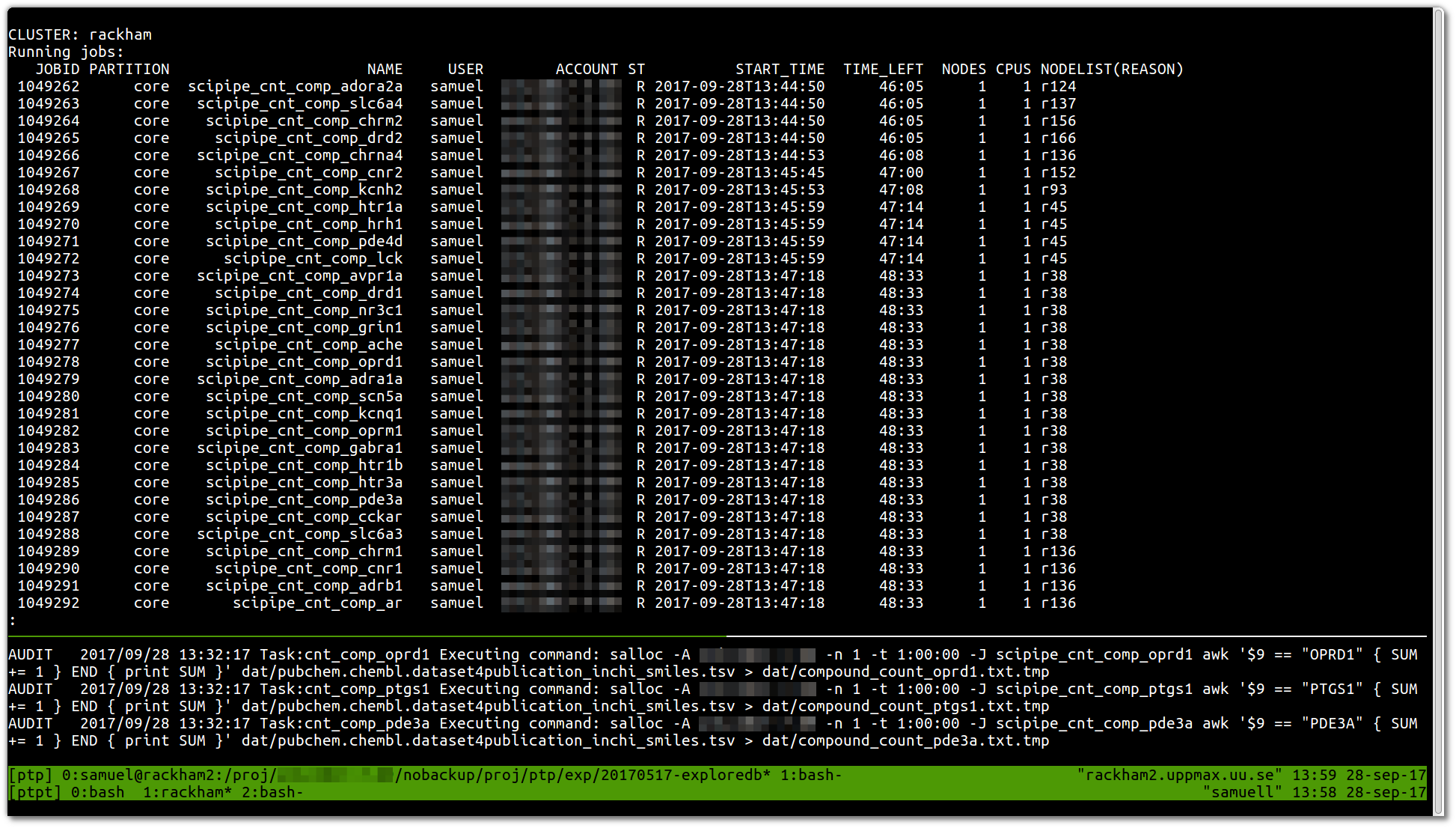
Today marked the day when we ran the very first production workflow with SciPipe , the Go -based scientific workflow tool we’ve been working on over the last couple of years. Yay! :)
This is how it looked (no fancy GUI or such yet, sorry):
The first result we got in this very very first job was a list of counts …
This post is also published on medium My current work at pharmb.io entails adding kubernetes support to my light-weight Go-based scientific workflow engine, scipipe (kubernetes, or k8s for short, is Google’s open source project for orchestrating container based compute clusters), which should take scipipe from a …
I’ve been following the development of D , Go and Rust (and also FreePascal for some use cases ) for some years (been into some benchmarking for bioinfo tasks ), and now we finally have three (four, with fpc) stable statically compiled languages with some momentum behind them, meaning they all are past 1.0. …

Some time ago I got a post published on GopherAcademy , outlining in detail how I think a flow-based programming inspired syntax can strongly help to create clearer, easier-to-maintain, and more declarative Go programs.
These ideas have since became clearer, and we (Ola Spjuth ’s research group at pharmbio ) …
I realize I didn’t have a link to my blog on Gopher Academy , on patterns for compoasable concurrent pipelines in Go(lang), so here it goes:
blog.gopheracademy.com/composable-pipelines-pattern
Edit: My original suggested way further below in the post is no way the “smallest pipeable” program, instead see this example (Credits: Axel Wagner ):
package main import ( "io" "os" ) func main() { io.Copy(os.Stdout, os.Stdin) } … or (credits: Roger Peppe ):
package main import ( …
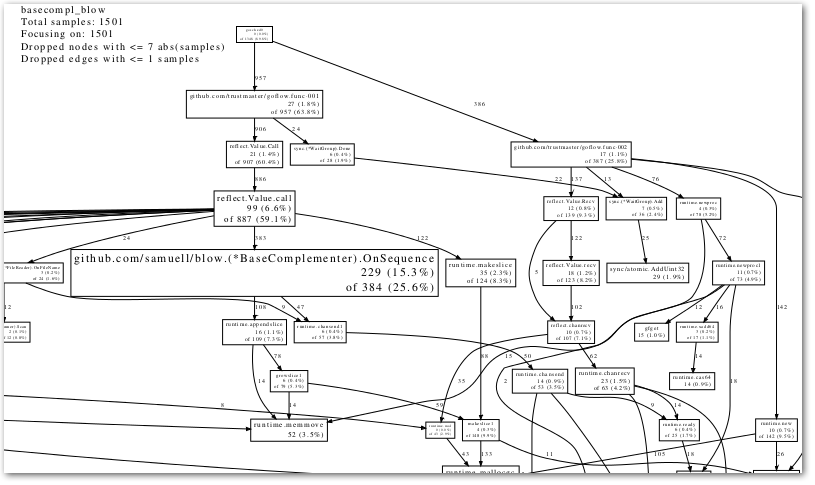
In trying to get my head around the code of the very interesting GoFlow library, (for flow-based programming in Go), and the accompanying flow-based bioinformatics library I started hacking on, I needed to get some kind of visualization (like a call graph) … something like this:
(And in the end, that is what I …