
Photo credits: Matthew Smith / Unsplash In our work on automating machine learning computations in cheminformatics with scientific workflow tools , we have came to realize something; Dynamic scheduling in scientific workflow tools is very important and sometimes badly needed.
What I mean is that new tasks should be …
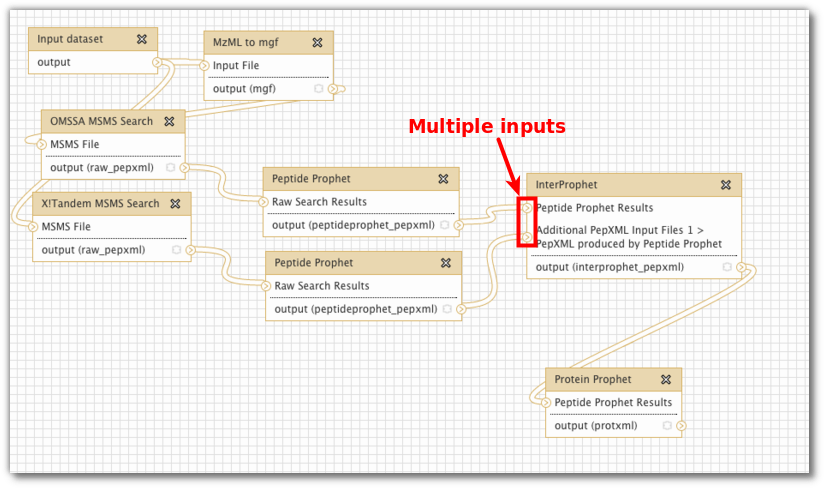
Upsurge in workflow tools There seem to be a little upsurge in light-weight - often python-based - workflow tools for data pipelines in the last couple of years: Spotify’s Luigi , OpenStack’s Mistral , Pinterest’s Pinball , and recently AirBnb’s Airflow , to name a few. These are all interesting …
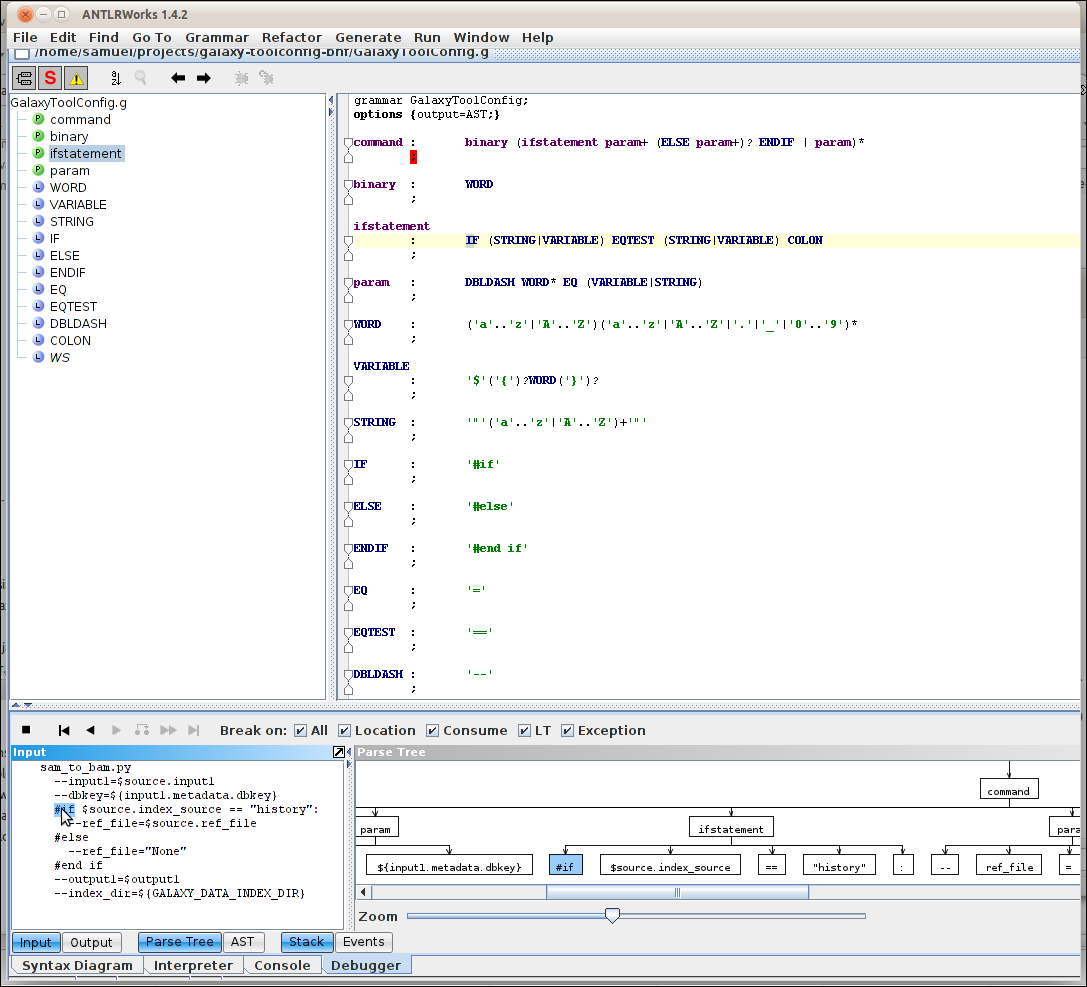
As blogged earlier, I’m currently into parsing the syntax of some definitions for the parameters and stuff of command line tools. As said in the linked blog post, I was pondering whether to use the Galaxy Toolconfig format or the DocBook CmdSynopsis format . It turned out though Well, that cmdsynopsis lacks the …
<tool id="sam_to_bam" name="SAM-to-BAM" version="1.1.1"> <description>converts SAM format to BAM format</description> <requirements> <requirement type="package">samtools</requirement> </requirements> <command interpreter="python"> …