Posts
In bioinformatics we are handling a lot of tabular data. Be it VCF files, tabular Blast output, or just creating a CSV or TSV samplesheet.
Actually, one of my favorite tabular formats is by using SeqKit to convert Fasta or FastQ files to tabular format, as this allows to do various filtering operations by row, using …
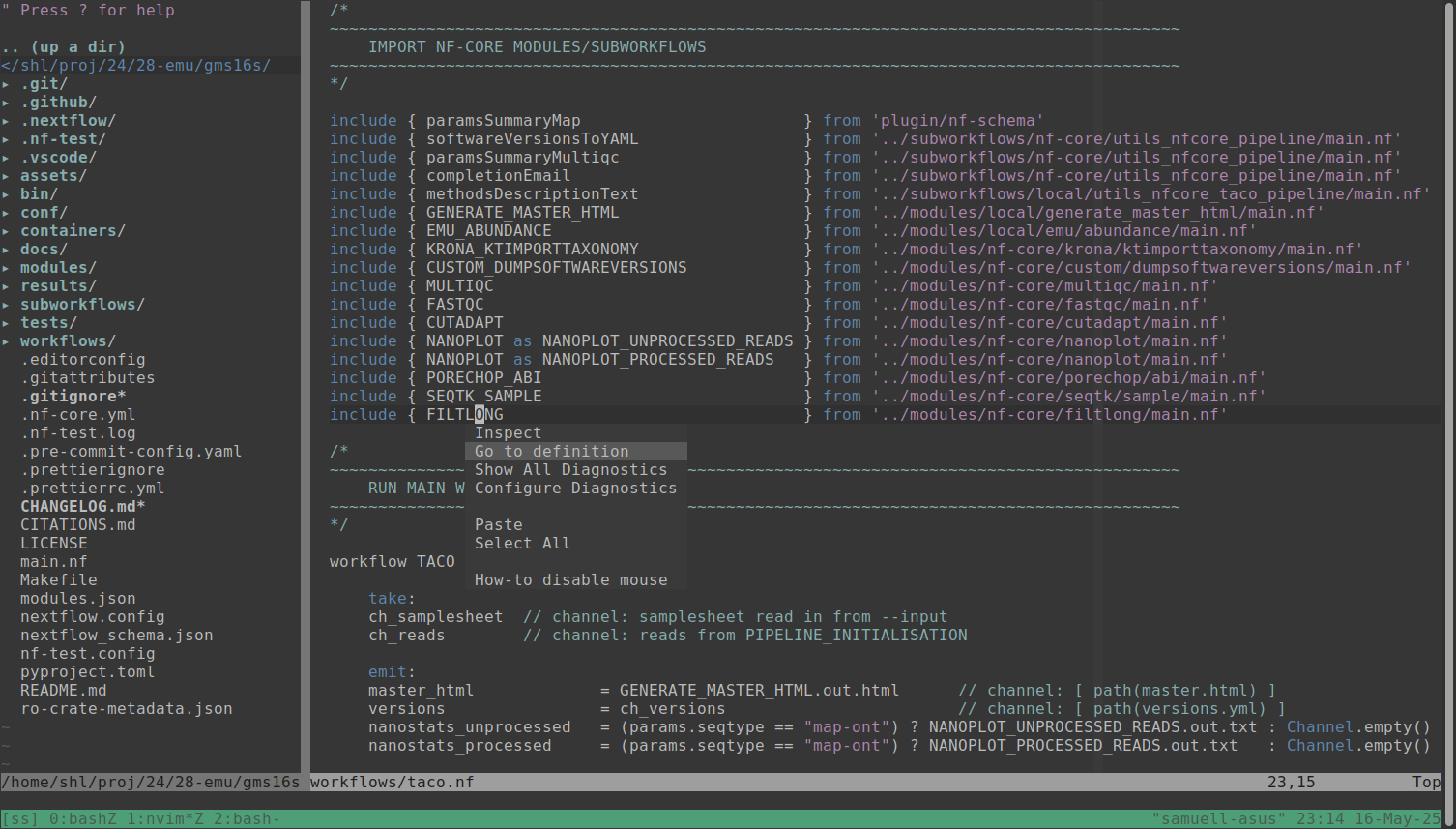
It turns out it is super easy to get Nextflow code intelligence to work in NeoVim now! This is thanks to the newly released Nextflow language server following the increasingly popular Language Server Protocol (LSP).
See this episode of the Nextflow podcast for a more in-depth discussion on this new functionality.
The …

Ever ran into a Linux box that just won’t boot, even after making sure that BIOS settings are OK and that no major hardware errors are at hand?
Then you need to know about the chroot technique, which can be a real life saver.
For example, I managed to repair a Nanopore GridION device this way the other week, …

I’m not the only one who thinks the R language can be pretty frustrating at times.
In fact, although I have been using it a dozen times over my career, every time I have picked it up again after a hiatus, I have been completely lost.
Many people have written about various aspects of why it is so confusing, and …

Bioinformatics is growing in the clinical field, and in my job in a clinical microbiology lab, I’m increasingly asked for tips about how to get into bioinformatics or genomic data science.
As I recently took the plunge into genomics from my PhD field of small molecular structures and machine learning, as part of …

When I presented a poster about SciCommander at the Swedish bioinformatics workshop last year, I got a lot of awesome feedback from some great people including Fredrik Boulund, Johannes Alneberg and others, of which I unfortunately lost the names (please shout out if you read this!).
(For those new to SciCommander, it …

I’m just back from the Applied Hologenomics Conference in 2024 (See also #AHC2024 on Twitter ) in Copenhagen and thought to reflect a little on the conference and highlight the bits that particularly stuck with me.
The first thing I want to say is that a paradigm shift is happening here.
I think what is …

Ad-hoc tasks in bioinformatics can contain such an immense number of operations and tasks that need to be performed to achieve a certain goal. Often these are all individually regarded as rather “standard” or “routine”. Despite this, it is quite hard to find an authoritative set of …

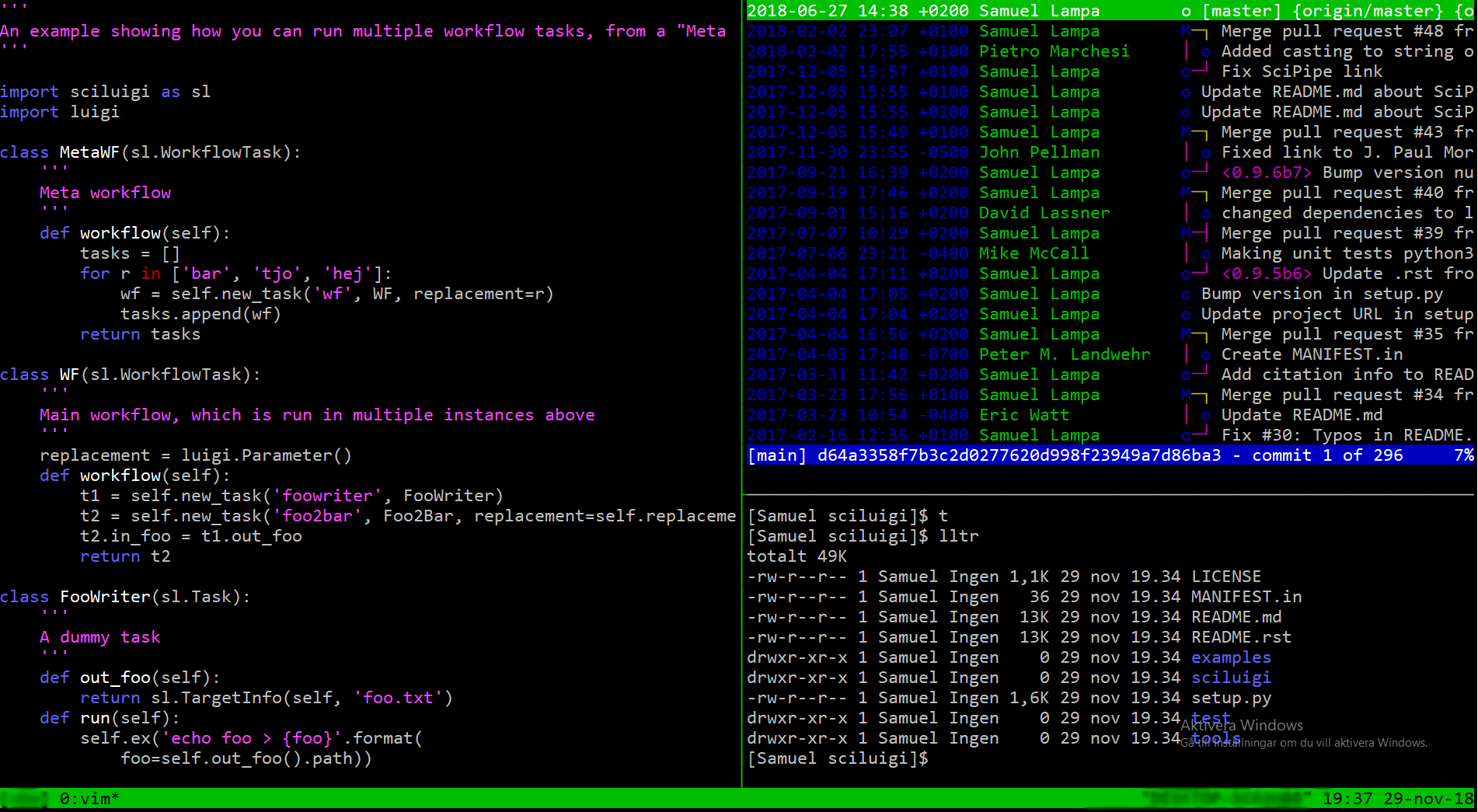
As we are - according to some expert opinions - living in the Century of Biology, I found it interesting to reflect on Go’s usage within the field.
Go has some great features that make it really well suited for biology, such as:
A relatively simple language that can be learned in a short time even for people …

I was happy to see the publication finally going online , of work done at NASA Glenn Research Center , where SciPipe has been used to process and track provenance of the analyses, “Modeling the impact of thoracic pressure on intracranial pressure”. I’ve known the work existed for a couple of years, …
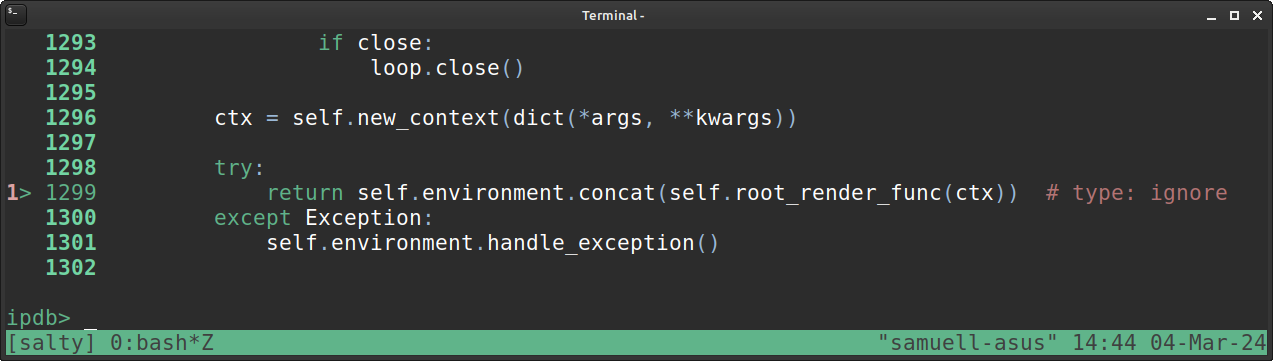
I’m working on a static reporting tool using the Jinja2 templating engine for Python.
I was trying to figure out a way to enter into the Jinja templating code with the pdb/ipdb commandline debugger.
I tried creating an .ipdbrc file in my local directory with the line:
path/to/template.html:<lineno> … …

I haven’t written much about a new tool I’ve been working on in some extra time: SciCommander .
I just presented a poster about it at the Swedish Bioinformatics Workshop 2023 , so perhaps let me first present you the poster instead of re-iterating what it is (click to view large version):
New version not …
We have been evaluating Nextflow before in my work at pharmb.io , but that was before DSL2 and the support for re-usable modules (which was one reason we needed to develop our own tools to support our challenges, as explained in the paper ). Thus, there’s definitely some stuff to get into.
Based on my years in …
See especially the end for info about how to set up a nice integration between the work and private accounts, such that one can e.g. occasionally start the mail client or web browser from the private account from the work one etc.
Caveats when installing Debian 11 Make sure that an EFI partition is created (when I …
I just switched to Qubes OS as operating system on my main work laptop (a Dell Latitude). Or in fact, one of the reasons was to be able to combine work and private hobby coding projects, that’s increasinbly been happening on the same machine. Anyways, these are my experiences and notes, as a way to document …
An area where I’m not so happy with some things I’ve seen in FP, is composability.
In my view, a well designed system or langauge should make functions (or other smallest unit of computation) more easily composable, not less.
What strikes me as one of the biggest elephants in the room regarding FP, is that …

I have been playing around a lot with concurrency in Go over the years, resulting in libraries such as SciPipe , FlowBase and rdf2smw . My main motivation for looking into Go has been the possibility to use it as a more performant, scaleable and type-safe alternative to Python for data heavy scripting tasks in …
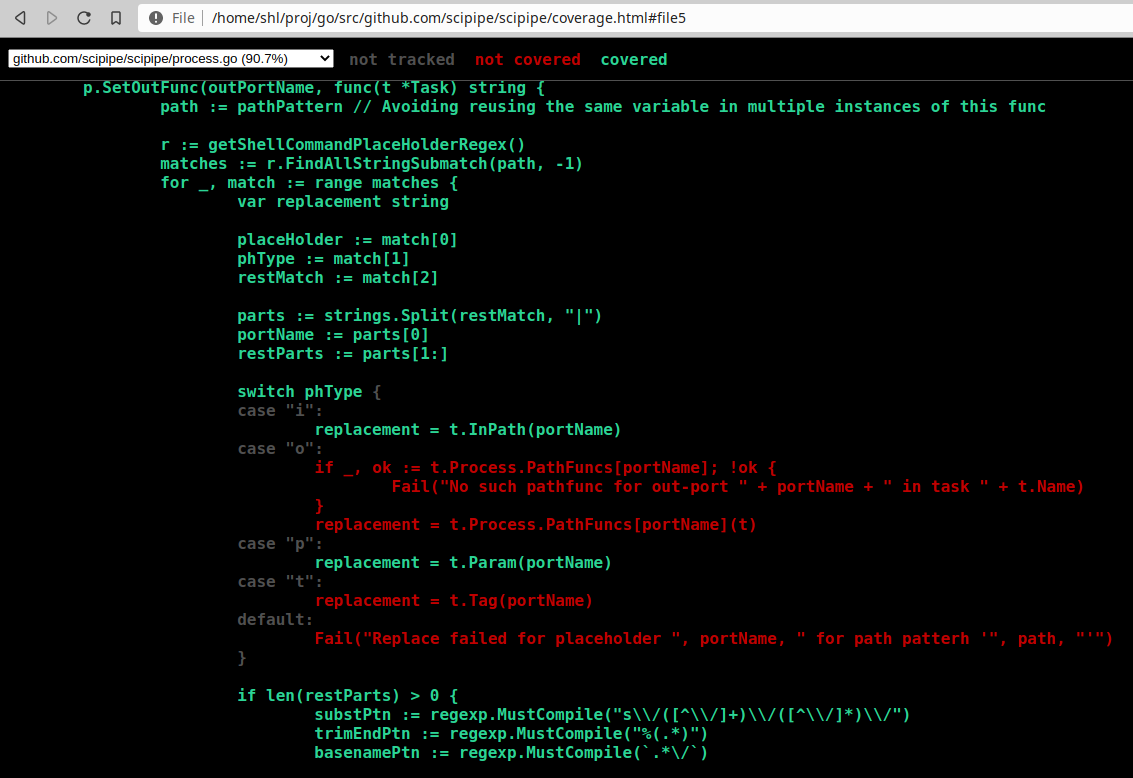
Go has some really nice tools for running tests and analyzing code. One of these functionalities is that you can generate coverage information when running tests, that can later be viewed in a browser using the go tool cover command. It turns out though, since doing it requires executing multiple commands after each …
wget -P . -mpck --html-extension -e robots=off --wait 0.5 <URL> To understand the flags, you can check man wget of course, but some explanations follow here:
-P - Tell where to store the site -m - Create a mirror -p - Download all the required files (.css, .js) needed to properly render the page -c - Continue …
ZeroMQ is a great way to quickly and simply send messages between multiple programs running on the same or different computers. It is very simple and robust since it doesn’t need any central server. Instead it talks directly between the programs through sockets, TCP-connections or similar.
ZeroMQ has client …
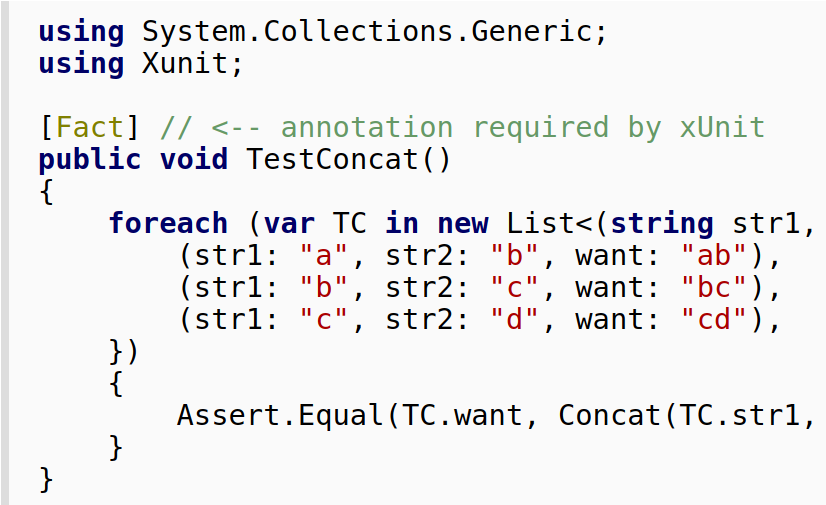
Folks in the Go community have championed so called table-driven tests (see e.g. this post by Dave Cheney and the Go wiki ) as a way to quickly and easily writing up a bunch of complete test cases with inputs and corresponding expected outputs, and looping over them to execute the function being tested. In short, the …

We just wanted to share that the paper on our Go-based workflow library, SciPipe, was just published in GigaScience:
Abstract Background The complex nature of biological data has driven the development of specialized software tools. Scientific workflow management systems simplify the assembly of such tools into …

I was so happy the other day to find someone else who found the great benefits of a little pattern for how to structure pipeline-heavy programs in Go, which I described in a few posts before. I have been surprised to not find more people using this kind of pattern, which has been so extremely helpful to us, so I …
I was looking for was a no-fuss, lightweight, robust and as simple as possible solution to running my normal Bash-based workflow inside the main Windows filesystem, interacting with the Windows world. Turns out there are some solutions. Read on for more info on that.
Windows Subsystem for Linux too heavy First, I must …
This is an excerpt from the “future outlook” section of my thesis titled “Reproducible Data Analysis in Drug Discovery with Scientific Workflows and the Semantic Web” (click for the open access full text), which aims to provide various putative ways towards improved reproducibility, …

A pre-print for our Go-based workflow libarary SciPipe , is out, with the title SciPipe - A workflow library for agile development of complex and dynamic bioinformatics pipelines , co-authored by me and colleagues at pharmb.io : Martin Dahlö , Jonathan Alvarsson and Ola Spjuth . Access it here .
It has been more than …
Update (May 2019): A paper incorporating the below considerations is published:
Björn A Grüning, Samuel Lampa, Marc Vaudel, Daniel Blankenberg, “Software engineering for scientific big data analysis ” GigaScience, Volume 8, Issue 5, May 2019, giz054, https://doi.org/10.1093/gigascience/giz054 There are a …
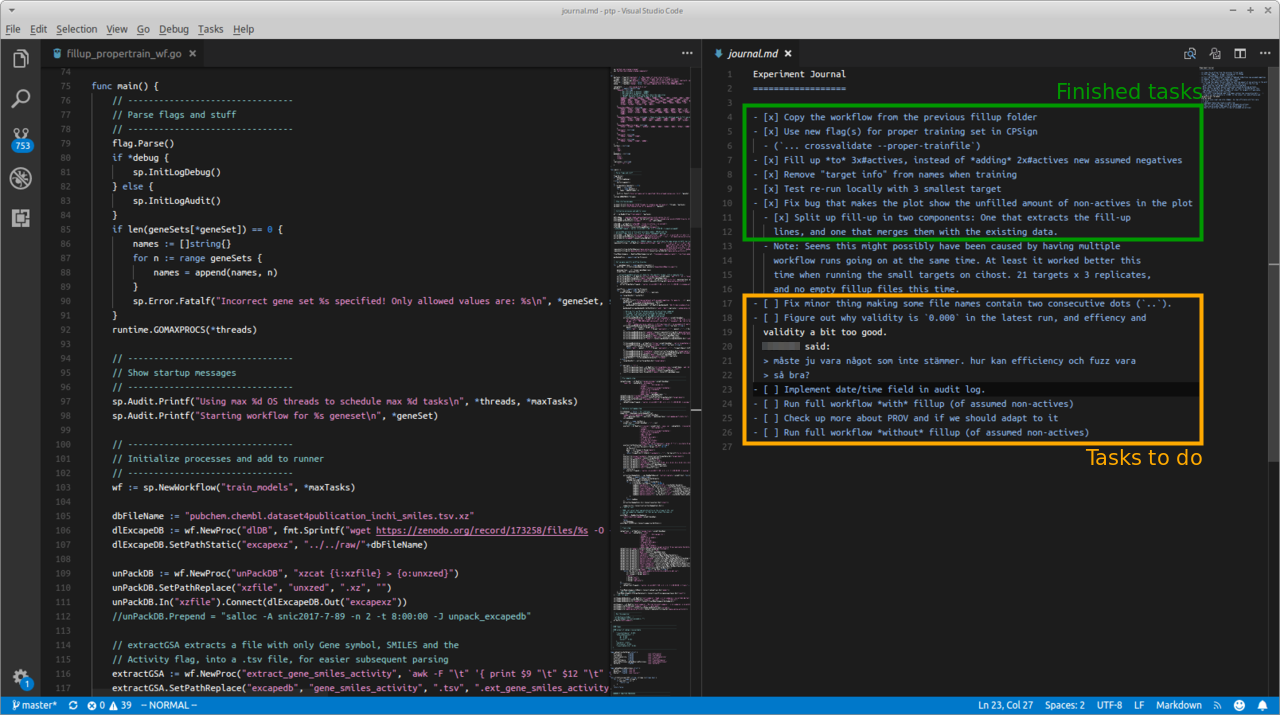
Good lab note-taking is hard Good note-taking is in my opinion as important for computational research as for wet lab research. For computational research it is much easier though to forget doing it, since you might not have a physical notebook lying on your desk staring at you, but rather might need to open a specific …
During the last months, I have had the pleasure work together with Matthias Palmér (MetaSolutions AB ) and Fernanda Dórea (National Veterinary Institute ), to prepare for and organize this year’s version of the annual Linked Data Sweden event , which this year was held in Uppsala hosted by the SciLifeLab Data …
I had a problem in which I thought I needed to parse the full DrugBank dataset, which comes as a (670MB) XML file (For open access papers describing DrugBank, see: [1], [2], [3] and [4]). It turned out what I needed was available as CSV files under “Structure External Links ”. There is probably still some …
Mathematical notation and dataflow programming Even though computations done on computers are very often based on some type of math, it is striking that the notation used in math to express equations and relations is not always very readily converted into programming code. Outside of purely symbolic programming …
Workflows and DAGs - Confusion about the concepts Jörgen Brandt tweeted a comment that got me thinking again on something I’ve pondered a lot lately:
“A workflow is a DAG.” is really a weak definition. That’s like saying “A love letter is a sequence of characters.” representation ≠ …

TL;DR: We wrote a post on gopherdata.io, about the growing ecosystem of Go-based workflow tools in bioinformatics. Go read it here It is interesting to note how Google’s Go programming language seems to increase in popularity in bioinformatics.
Just to give a sample of some of the Go based bioinformatics tools …
One year left to the dissertation (we hope) and now turning from mostly software development into more of data analysis and needing to read up quite a pile of books and papers on my actual topic, pharmaceutical bioinformatics. With this background, I’m feel forced to ponder ways to improving my note taking …

I’m reading A mind for numbers , by Barbara Oakley. Firstly, it is a very interesting book, but the main lesson I’ve already learned from this book seems so paramount that I have to write it down, so I don’t forget it (some meta-connotations in that statement ;) ). I found the book through …
One of the more important tasks for a scientific workflow is to keep track of so called “provenance information” about its data outputs - information about how each data file was created. This is important so other researchers can easily replicate the study (re-run it with the same software and tools). It …
Thus, optimally, one would want to use Go’s handy range keyword for looping over multiple channels, since range takes care of closing the for-loop at the right time (when the inbound channel is closed). So something like this (N.B: non-working code!):
for a, b, c := range chA, chB, chC { doSomething(a, b, c) } …
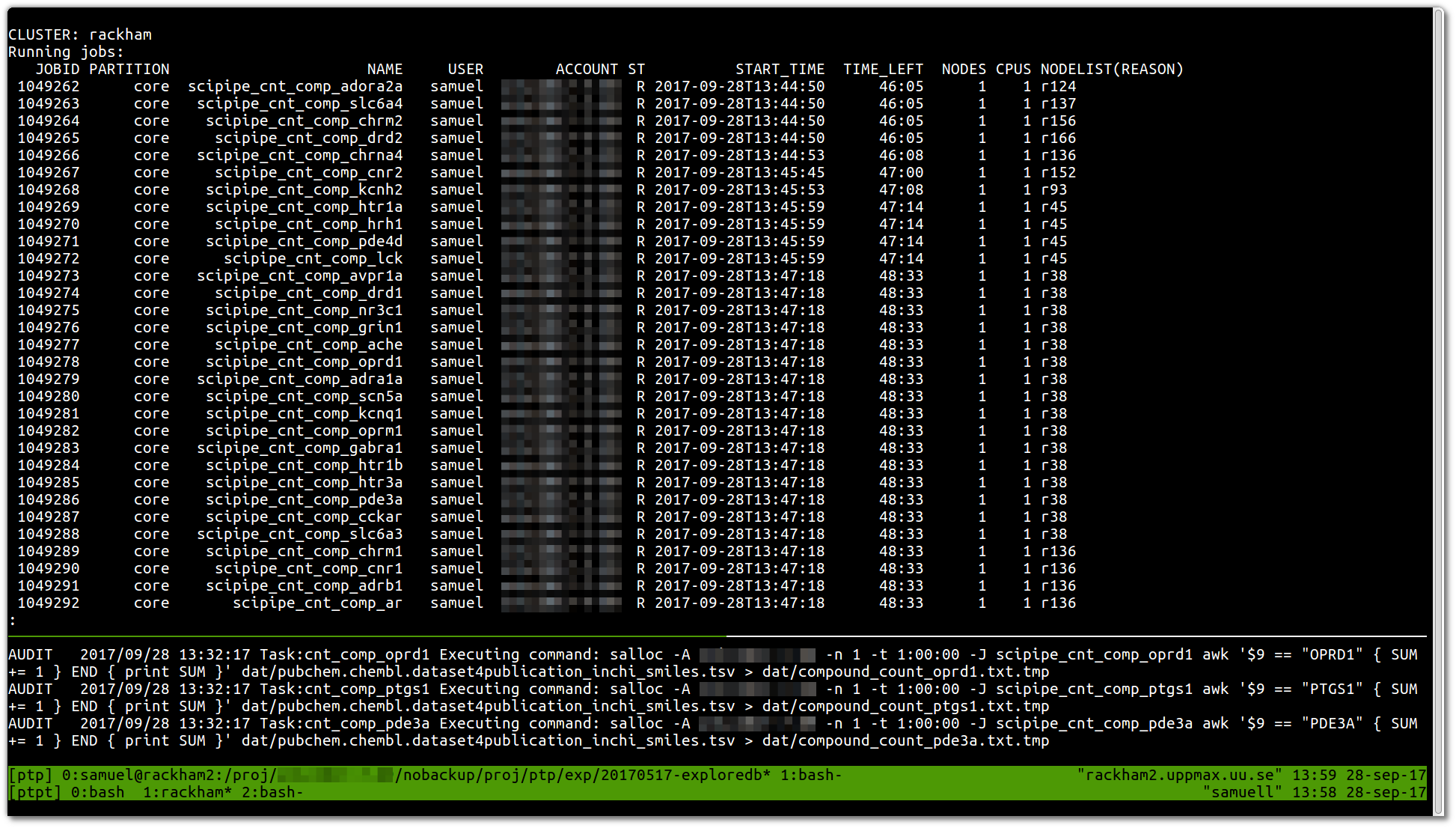
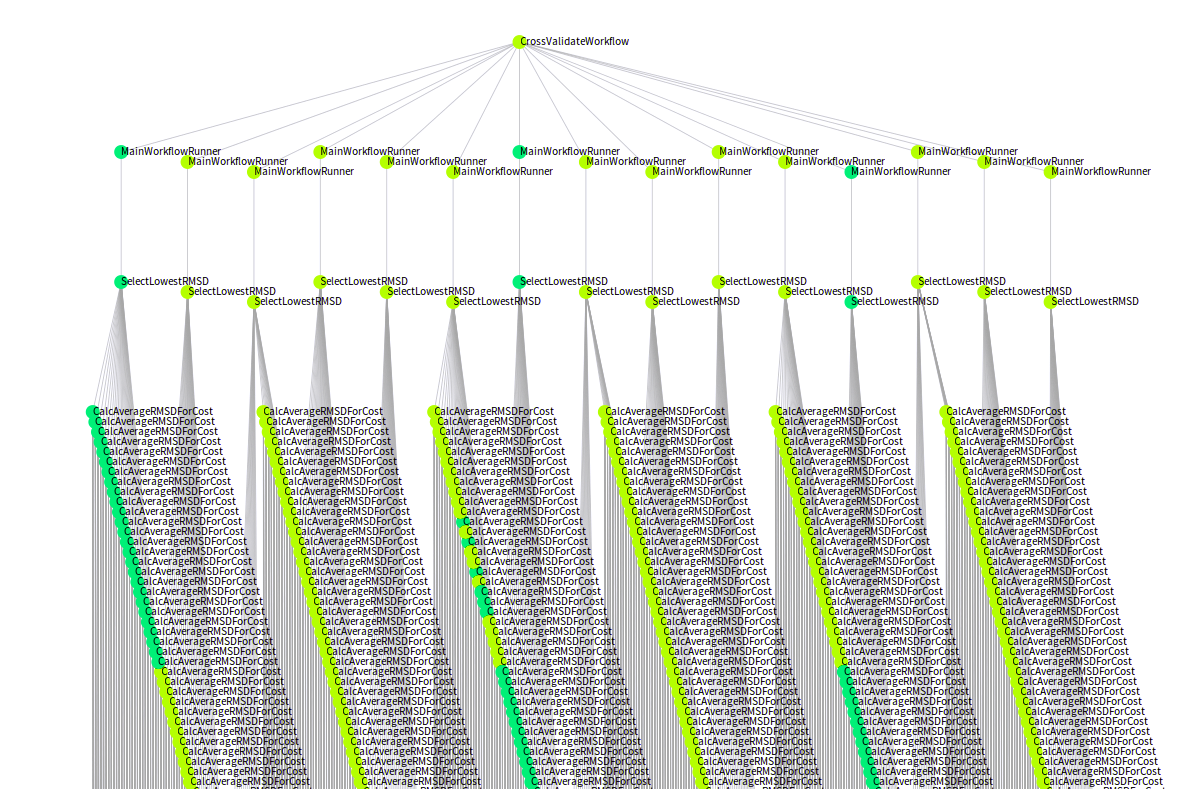
Today marked the day when we ran the very first production workflow with SciPipe , the Go -based scientific workflow tool we’ve been working on over the last couple of years. Yay! :)
This is how it looked (no fancy GUI or such yet, sorry):
The first result we got in this very very first job was a list of counts …
At pharmb.io we are researching how to use semantic technologies to push the boundaries for what can be done with intelligent data processing, often of large datasets (see e.g. our paper on linking RDF to cheminformatics and proteomics , and our work on the RDFIO software suite ). Thus, for us, RDFHDT opens new …
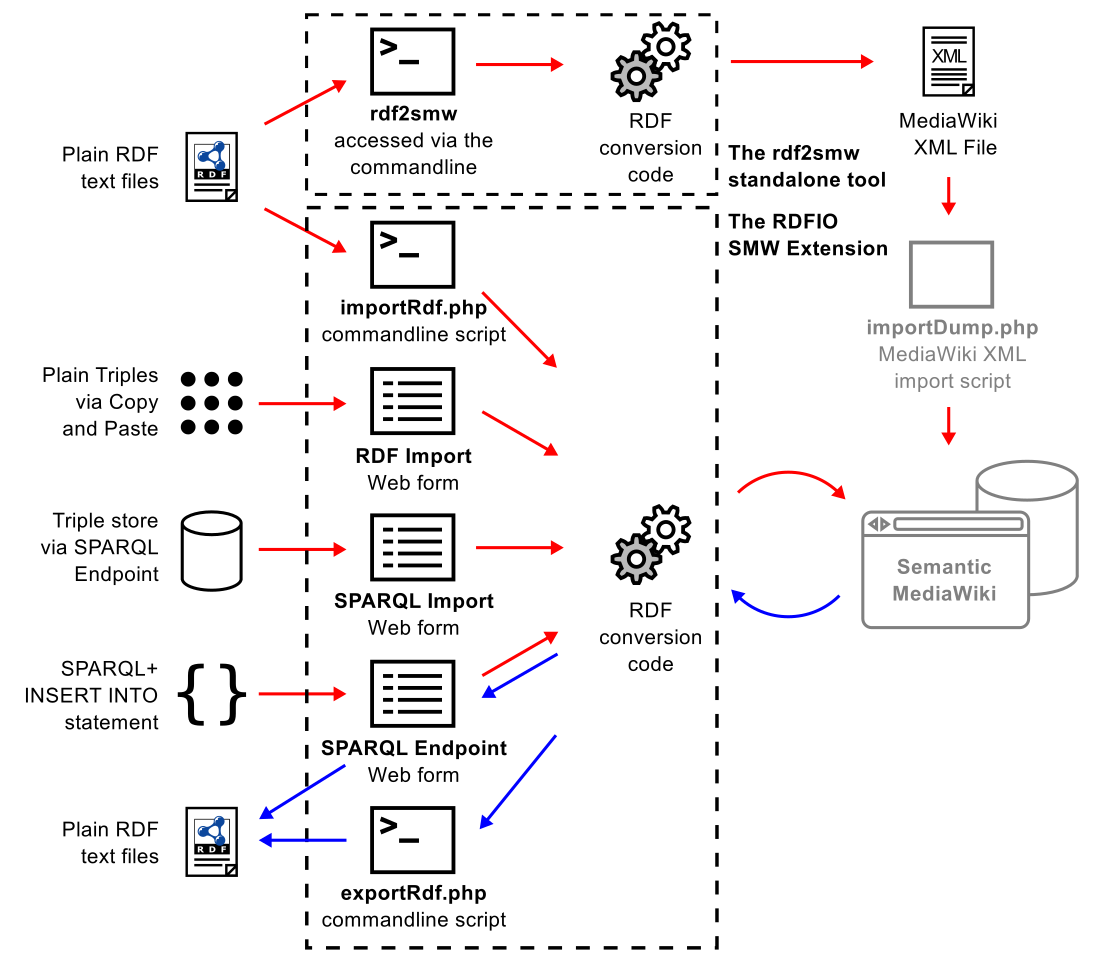
As my collaborator and M.Sc. supervisor Egon Willighagen already blogged , we just released a paper titled: “RDFIO: extending Semantic MediaWiki for interoperable biomedical data management ”, with uses cases from Egon and Pekka Kohonen , coding help from Ali King and project supervision from Denny …
This post is also published on medium My current work at pharmb.io entails adding kubernetes support to my light-weight Go-based scientific workflow engine, scipipe (kubernetes, or k8s for short, is Google’s open source project for orchestrating container based compute clusters), which should take scipipe from a …

I was invited to give a talk at Semantic MediaWiki (SMW) conference in Frankfurt last week, on our work on enabling import of RDF datasets into SMW . I have presented at SMWCon before as well (2011: blog , slides video , 2013: slides ), so it was nice to re-connect with some old friends, and to get up to date about how …
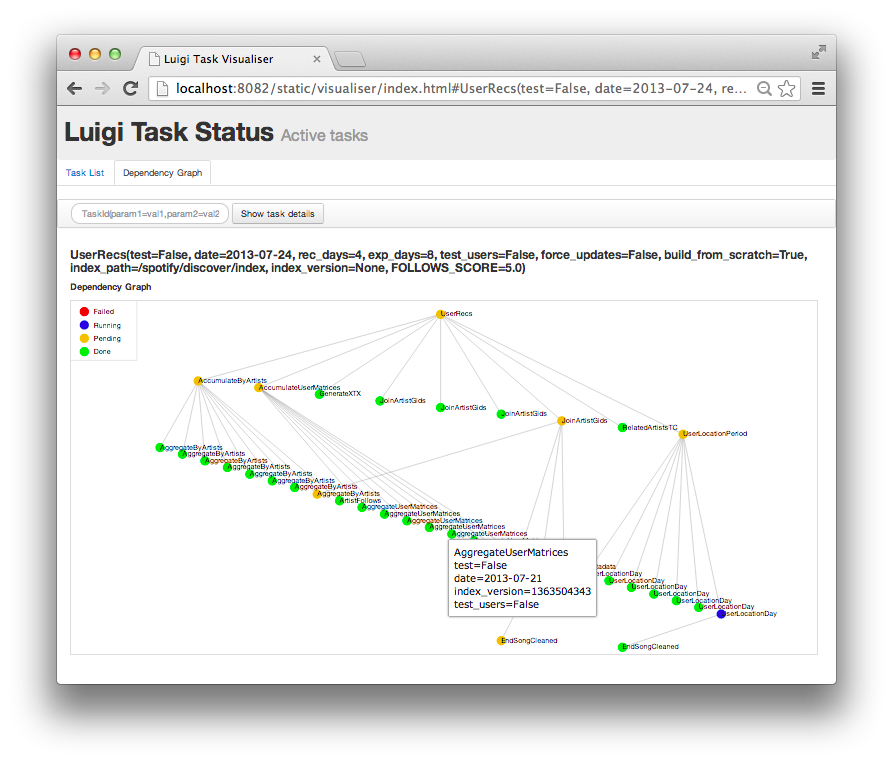
This is a Luigi tutorial I held at the e-Infrastructures for Massively parallel sequencing workshop (Video archive ) at SciLifeLab Uppsala in January 2015, moved here for future reference.
What is Luigi? Luigi is a batch workflow system written in Python and developed by Erik Bernhardson and others at Spotify , where …
I’ve been following the development of D , Go and Rust (and also FreePascal for some use cases ) for some years (been into some benchmarking for bioinfo tasks ), and now we finally have three (four, with fpc) stable statically compiled languages with some momentum behind them, meaning they all are past 1.0. …
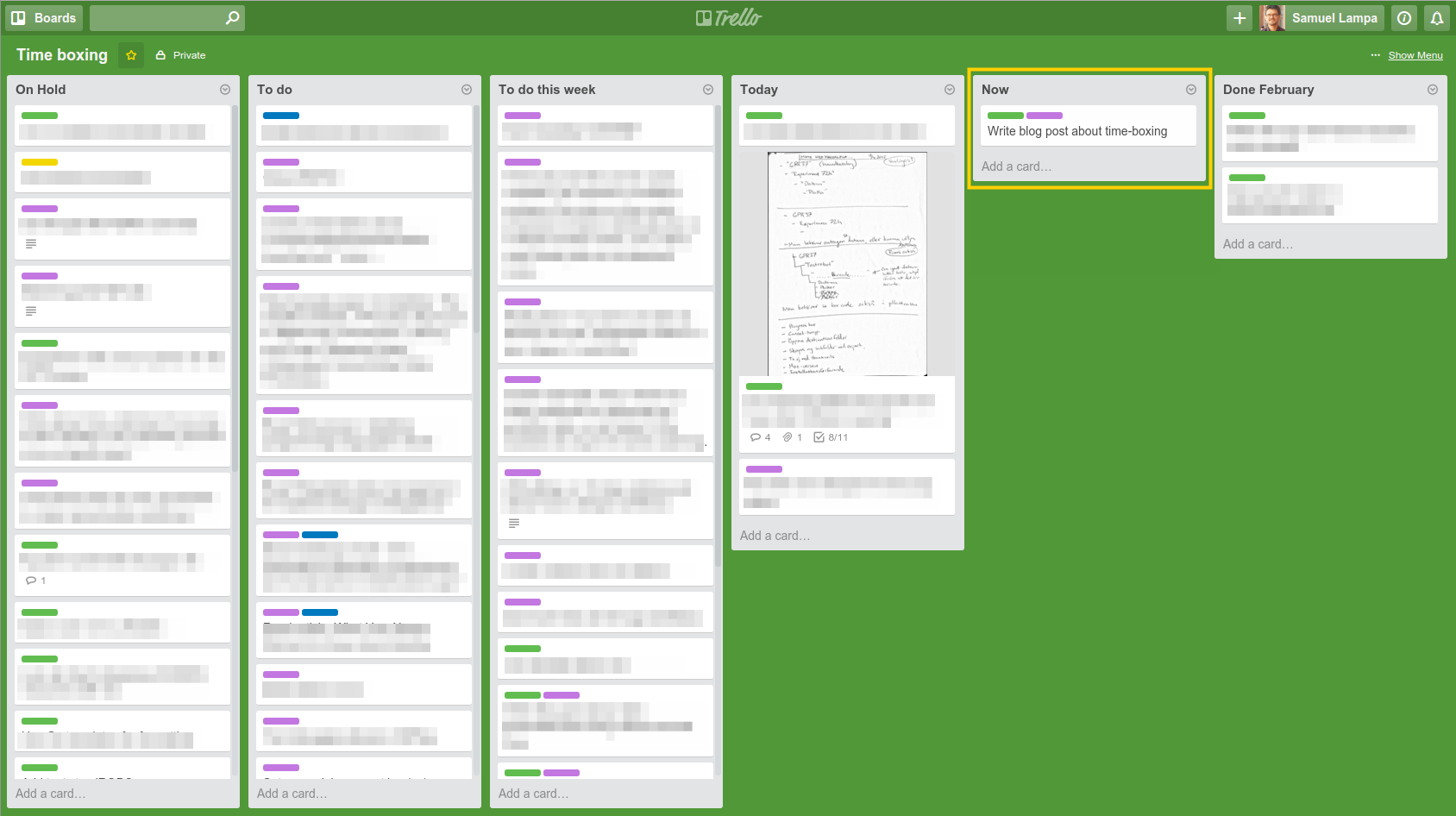
Figure: Sketchy screenshot of how my current board looks. Notice especially the “Now” stack, marked in yellow, where you are only allowed to put one single card. I used to have a very hard time getting an overview of my current work, and prioritizing and concentrating on any single task for too long. I …

I was working with a migration from drupal to processwire CMS:es, where I wanted to be able to pipe data, including the body field with HTML formatting and all, through multiple processing steps in a flexible manner. I’d start with an extraction SQL query, through a few components to replace and massage the data, …
After just reading on Hacker News about Google’s newly released TensorFlow library , for deep learning based on tensors and data flow, I realized I wrote in a draft post back in 2013 that:
“What if one could have a fully declarative “matrix language” in which all data transformations ever needed …

Photo credits: Matthew Smith / Unsplash In our work on automating machine learning computations in cheminformatics with scientific workflow tools , we have came to realize something; Dynamic scheduling in scientific workflow tools is very important and sometimes badly needed.
What I mean is that new tasks should be …

I had heard a lot of people say vim is very hard to learn, and got the impression that it will take a great investment to switch to using it.
While I have came to understand that they are right in that there is a lot of things to invest in to get really great at using vim, that will really pay back, I have also found …
This manifested itself in a bunch of error message from the python module in vim, ending with:
AttributeError: 'module' object has no attribute 'vars' I first thought it was an error in vim-pyenv and reported it (see that issue for more in-depth details). In summary it turns out that older versions of …

Some time ago I got a post published on GopherAcademy , outlining in detail how I think a flow-based programming inspired syntax can strongly help to create clearer, easier-to-maintain, and more declarative Go programs.
These ideas have since became clearer, and we (Ola Spjuth ’s research group at pharmbio ) …
I have tried hard to improve my linux desktop productivity by learning to do as much as possible using keyboard shortcuts, aliases for terminal commands etc etc (I even produced an online course on linux commandline productivity ).
In this spirit, I naturally tried out a so called tiling window manager (aka tiling wm). …

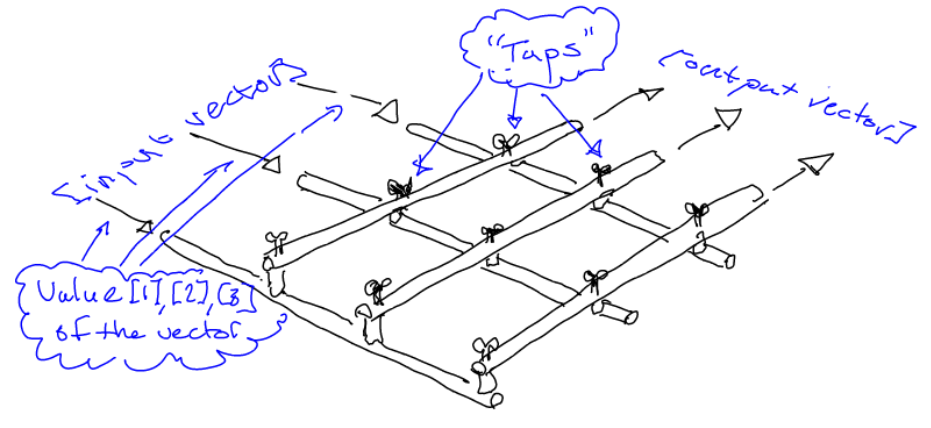
Often when I suggest people have a look at Flow-based Programming (FBP) or Data Flow for one reason or another, people are often put off by the strong connection between these concepts and graphical programming. That is, the idea that programs will be easier to understand if expressed and developed in a visual …
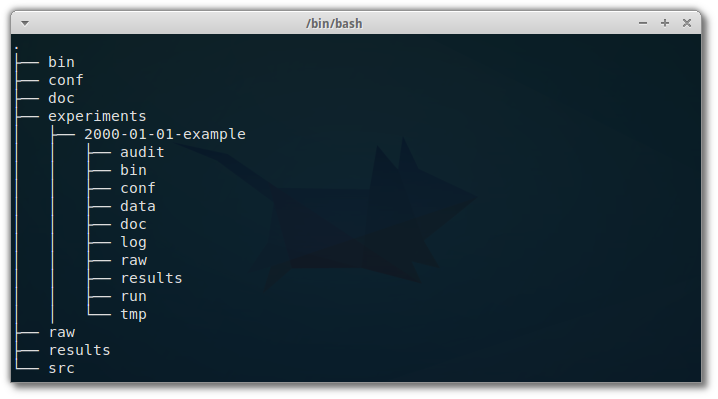
I read this excellent article with practical recommendations on how to organize a computational project, in terms of directory structure.
Directory structure matters The importance of a good directory structure seems to often be overlooked in teaching about computational biology, but can be the difference between a …

I think Erlang/Elixir fits great as control plane or service-to-service messaging layer for distributing services built with flow-based programming
Just back from a one day visit to Erlang User Conference . I find the Erlang virtual machine fascinating. And with the new Elixir language built on top of it to fix some of …

iRODS , the “integrated rule oriented data system” is a super cool system for managing datasets consisting of files, from smallish ones, to really large ones counted in petabytes, and possibly spanning multiple continents.
There’s a lot to be said about iRODS (up for another blog post) but the one …
Upsurge in workflow tools There seem to be a little upsurge in light-weight - often python-based - workflow tools for data pipelines in the last couple of years: Spotify’s Luigi , OpenStack’s Mistral , Pinterest’s Pinball , and recently AirBnb’s Airflow , to name a few. These are all interesting …
I realize I didn’t have a link to my blog on Gopher Academy , on patterns for compoasable concurrent pipelines in Go(lang), so here it goes:
blog.gopheracademy.com/composable-pipelines-pattern

Disclaimer: Don’t take this too seriously … this is “thinking-in-progress” :)
It just struck me the other minute, how simplicity is the key theme behind two very important areas in software development, that I’ve been dabbling with quite a bit recently: Testing, and automation.
Have you …
The workflow problem solved once and for all in 1979? As soon as the topic of scientific workflows is brought up, there are always a few make fans fervently insisting that the problem of workflows is solved once and for all with GNU make , written first in the 70’s :)
Personally I haven’t been so sure. On …
Improving performance in Delphi Bold MDA applications by replacing navigation code with derived links in the model This post on Model Driven Architecture in Delphi and Bold , by Rolf Lampa , has been previously published on howtodothings.com .
Modeling class structures takes some thinking, and when done the thinking …

It turned out I didn’t have the time and strength to blog every day at the NGS Bioinformatics Intro course, so here comes a wrap up with some random notes and tidbits from the last days, including any concluding remarks!
These days we started working on a more realistic NGS pipeline, on analysing re-sequencing …
Some random links from the Hadoop for Next-Gen Sequencing workshop held at KTH in Kista, Stockholm in February 2015.
UPDATE: Slides and Videos now available !
Spark notebook Scala notebook ADAM By Big Data Genomics Tweet by Frank Nothaft on common workflow def Part of Global Alliance for … Another link is …
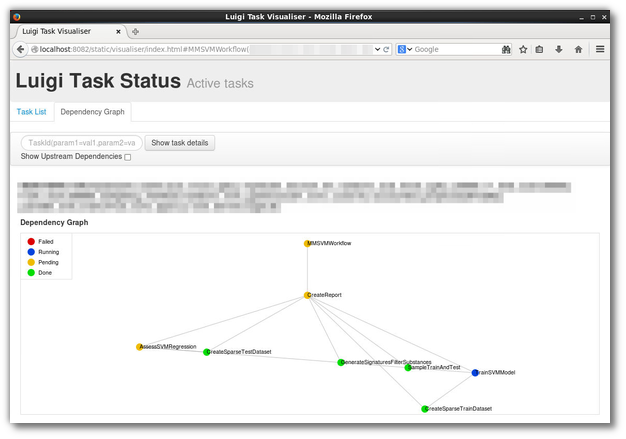
Fig 1: A screenshot of Luigi’s web UI, of a real-world (although rather simple) workflow implemented in Luigi:
Update May 5, 2016: Most of the below material is more or less outdated. Our latest work has resulted in the SciLuigi helper library , which we have used in production and will be focus of further …

Today was the second day of the introductory course in NGS bioinformatics that I’m taking as part of my PhD studies.
For me it started with a substantial oversleep, probably due to a combination of an annoying cold and the ~2 hour commute from south Stockholm to Uppsala and BMC . Thus I missed some really …

Just finished day 1 of the introductory course on Bioinformatics for Next generation sequencing data at Scilifelab Uppsala. Attaching a photo from one of the hands-on tutorial sessions, with the tutorial leaders, standing to the right.
Today’s content was mostly introductions to the linux commandline in general, …
Right now I’m sitting on the train and trying to get my head around some of the pre-course materials .
The old Virtual Machine still available The old virtual machine from June 25, 2014, based on Ubuntu 14.04, and RDFIO 2.x can be found here
Edit: My original suggested way further below in the post is no way the “smallest pipeable” program, instead see this example (Credits: Axel Wagner ):
package main import ( "io" "os" ) func main() { io.Copy(os.Stdout, os.Stdin) } … or (credits: Roger Peppe ):
package main import ( …
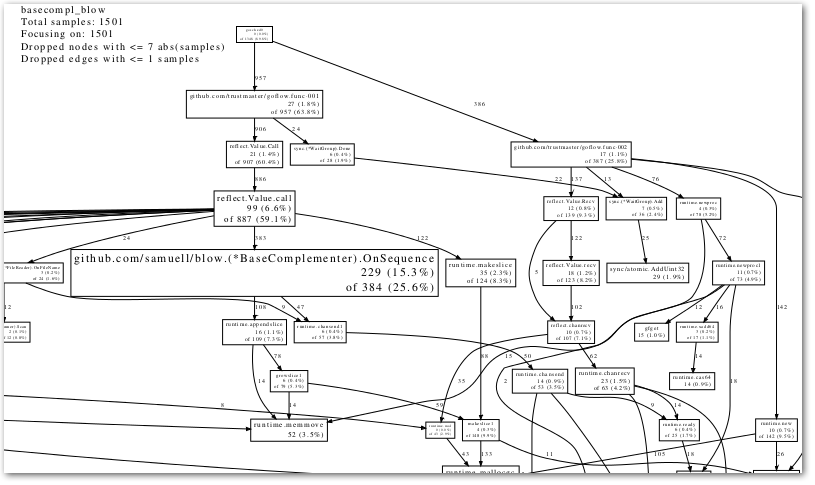
In trying to get my head around the code of the very interesting GoFlow library, (for flow-based programming in Go), and the accompanying flow-based bioinformatics library I started hacking on, I needed to get some kind of visualization (like a call graph) … something like this:
(And in the end, that is what I …
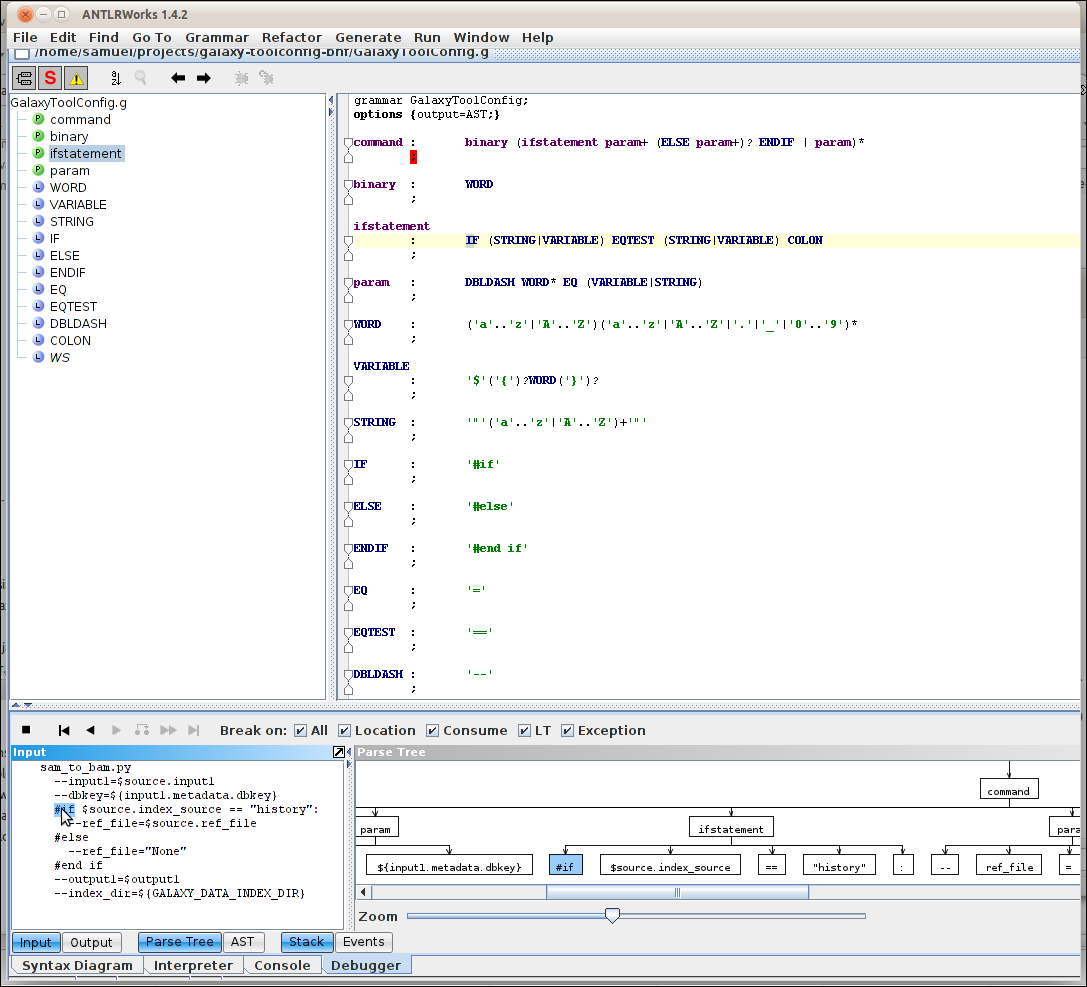
As blogged earlier, I’m currently into parsing the syntax of some definitions for the parameters and stuff of command line tools. As said in the linked blog post, I was pondering whether to use the Galaxy Toolconfig format or the DocBook CmdSynopsis format . It turned out though Well, that cmdsynopsis lacks the …
<tool id="sam_to_bam" name="SAM-to-BAM" version="1.1.1"> <description>converts SAM format to BAM format</description> <requirements> <requirement type="package">samtools</requirement> </requirements> <command interpreter="python"> …
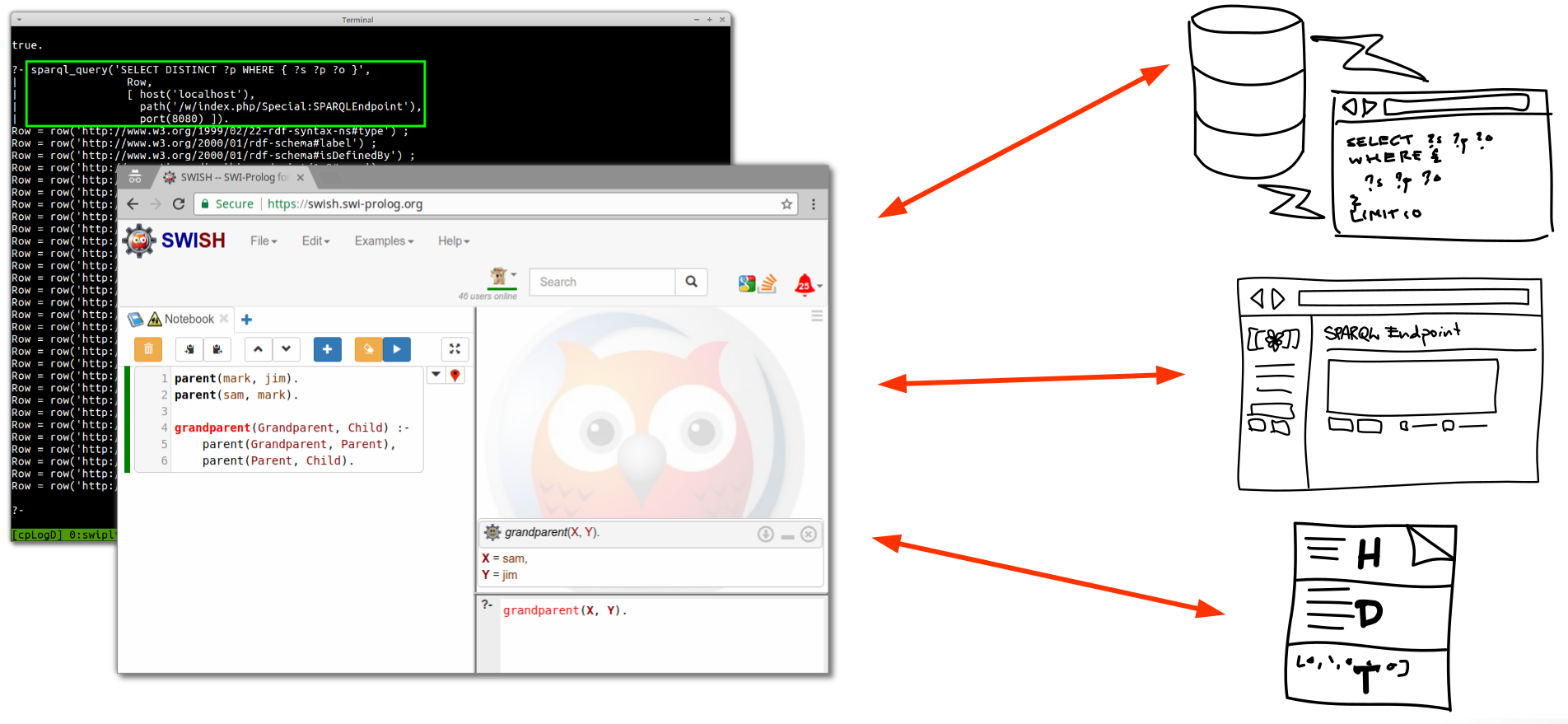
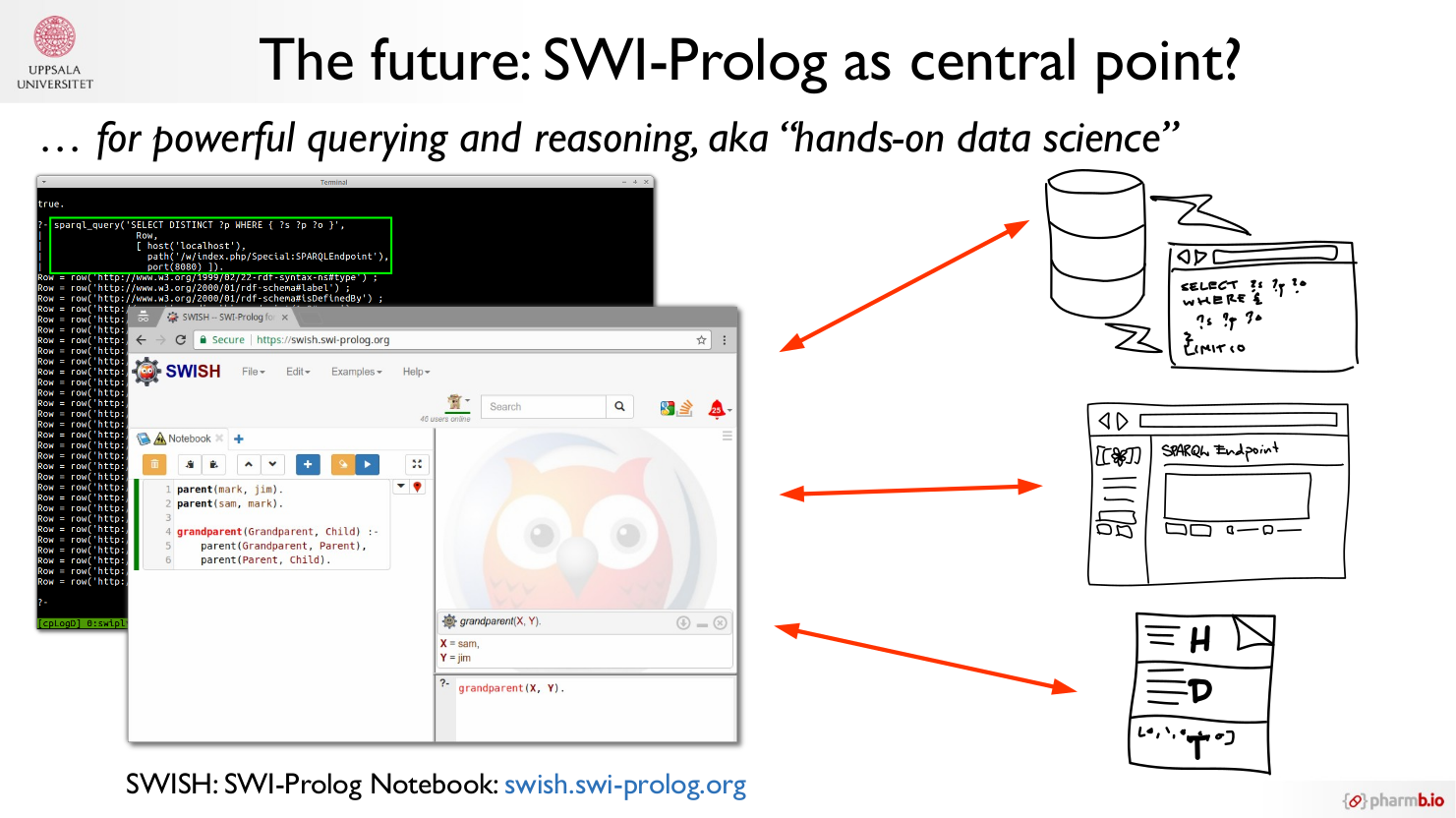
There are lots of things that can’t be answered by a computer from data alone. Maybe the majority of what we humans perceive as knowledge is inferred from a combination of data (simple fact statements about reality) and rules that tell how facts can be combined together to allow making implicit knowledge …
Then you can load the program from inside prolog after you’ve started it.
So, let’s start the prolog interactive GUI:
prolog Then, in the Prolog GUI, load the file test.pl like so:
?- [test]. Now, if you had some prolog clauses in the test.pl file, you will be able to extract that information by querying. …